
Inactivation of the Akt/FOXM1 Signaling Pathway by Panobinostat Suppresses the Proliferation and Metastasis of Gastric Cancer Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Inhibition of Cell Growth and Proliferation by Panobinostat
2.2. Induction of Apoptosis by Panobinostat
2.3. Cell Cycle Regulation by Panobinostat
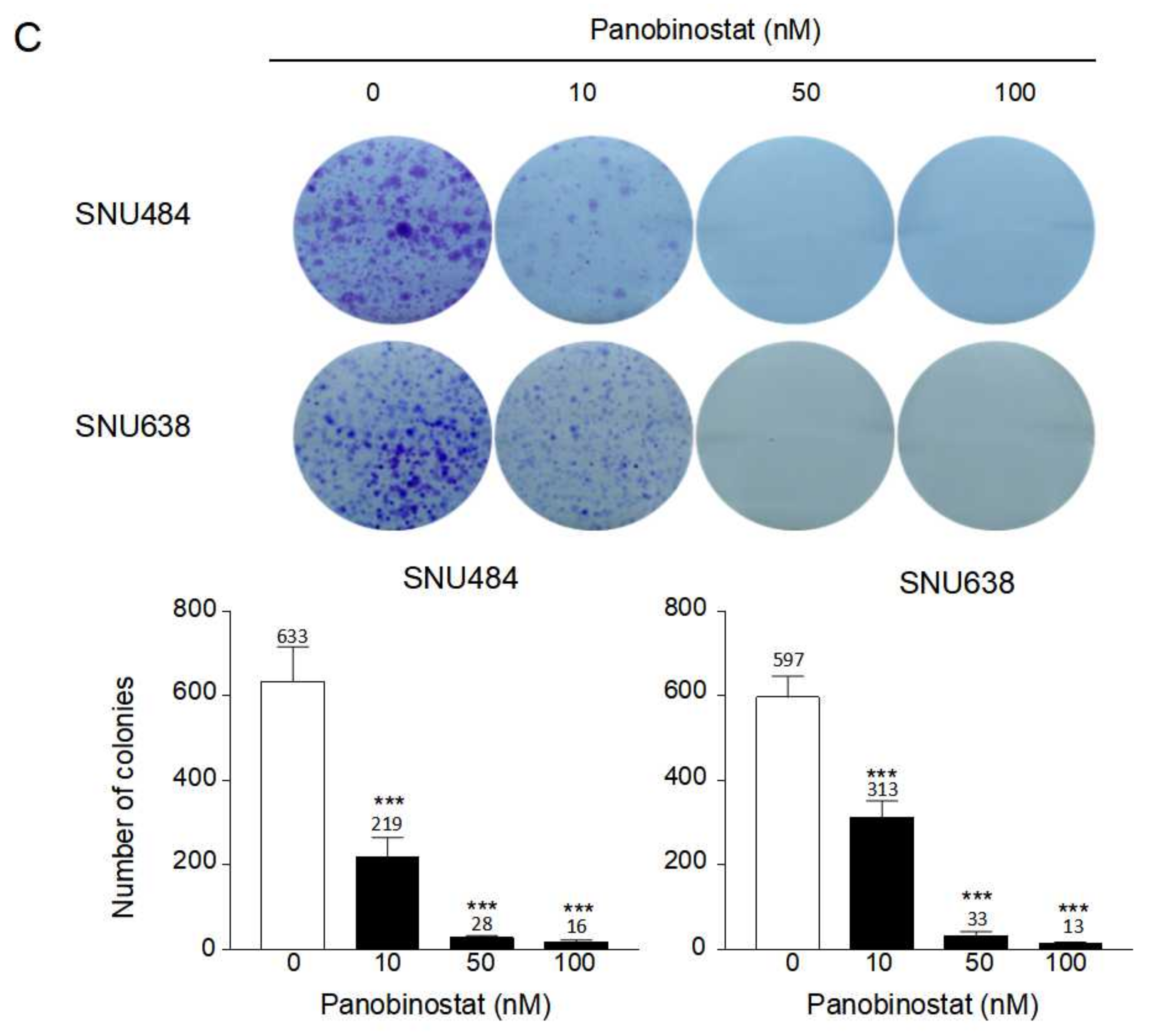
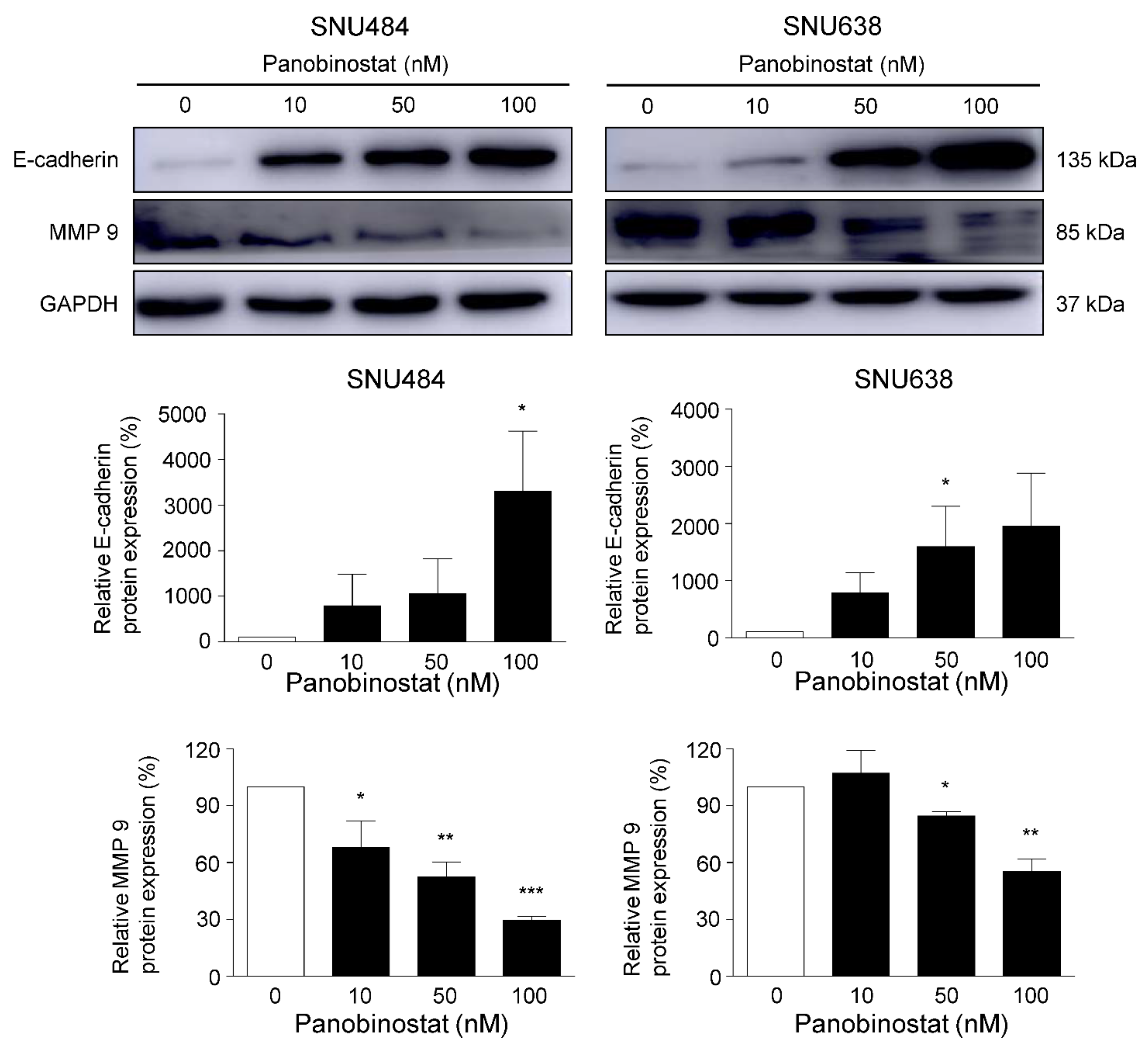
2.4. Cell Migration and Metastasis Inhibition by Panobinostat
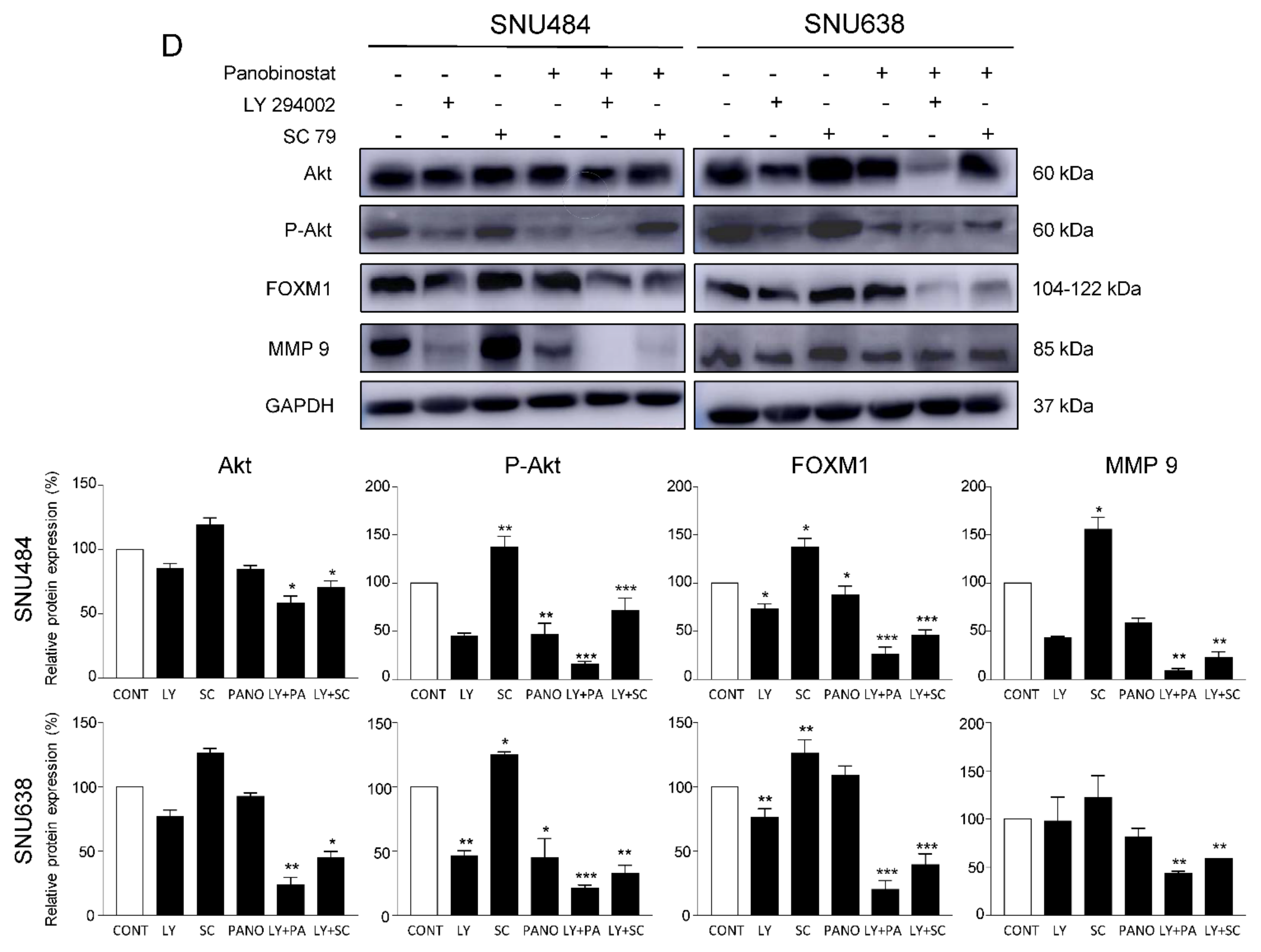
2.5. Downregulation of Akt/FOXM1 Signaling by Panobinostat
2.6. Inhibitory Effect of Panobinostat on Tumor Growth in an In Vivo Xenograft Animal Model
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Cell Viability Assay by WST Assay
4.3. Colony Formation Assay
4.4. Cell Cycle Analysis
4.5. Annexin V–Fluorescein Isothiocyanate (FITC) Staining Assay
4.6. Western Blot Analysis
4.7. Wound Healing Assay
4.8. Xenograft Experiment
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, D.; Chua, Y.J. East Meets West in the Treatment of Gastric Cancer. N. Engl. J. Med. 2007, 357, 1863–1865. [Google Scholar] [CrossRef]
- Macdonald, J.S. Gastric cancer-new therapeutic options. N. Engl. J. Med. 2006, 355, 76–77. [Google Scholar] [CrossRef] [Green Version]
- Wagner, J.M.; Hackanson, B.; Lübbert, M.; Jung, M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin. Epigenet. 2010, 1, 117–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef]
- Lee, H.-Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Shen, Y.L.; Chen, X.-H. FDA approval: Belinostat for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prince, H.M.; Bishton, M.J.; Johnstone, R.W. Panobinostat (LBH589): A potent pan-deacetylase inhibitor with promising activity against hematologic and solid tumors. Future Oncol. 2009, 5, 601–612. [Google Scholar] [CrossRef]
- Raedler, L.A. Farydak (Panobinostat): First HDAC inhibitor approved for patients with relapsed multiple myeloma. Am. Health Drug Benefits 2016, 9, 84–87. [Google Scholar]
- Healio. FDA approves Farydak for treatment of multiple myeloma. HemOnc Today, 23 February 2015. Available online: https://www.healio.com/news/hematology-oncology/20150223/fda-approves-farydak-for-multiple-myeloma (accessed on 30 April 2021).
- Tzogani, K.; van Hennik, P.; Walsh, I.; De Graeff, P.; Folin, A.; Sjoberg, J.; Salmonson, T.; Bergh, J.; Laane, E.; Ludwig, H.; et al. EMA Review of Panobinostat (Farydak) for the Treatment of Adult Patients with Relapsed and/or Refractory Multiple Myeloma. Oncologist 2018, 23, 631–636. [Google Scholar] [CrossRef] [Green Version]
- Tate, C.R.; Rhodes, L.V.; Segar, H.C.; Driver, J.L.; Pounder, F.N.; Burow, M.E.; Collins-Burow, B.M. Targeting triple-negative breast cancer cells with the histone deacetylase inhibitor panobinostat. Breast Cancer Res. 2012, 14, R79. [Google Scholar] [CrossRef] [Green Version]
- Crisanti, M.C.; Wallace, A.F.; Kapoor, V.; Vandermeers, F.; Dowling, M.L.; Pereira, L.P.; Coleman, K.; Campling, B.G.; Fridlender, Z.G.; Kao, G.D. The HDAC inhibitor panobinostat (LBH589) inhibits mesothelioma and lung cancer cells in vitro and in vivo with particular efficacy for small cell lung cancer. Mol. Cancer Ther. 2009, 8, 2221–2231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathkopf, D.; Wong, B.Y.; Ross, R.W.; Anand, A.; Tanaka, E.; Woo, M.M.; Hu, J.; Dzik-Jurasz, A.; Yang, W.; Scher, H.I. A phase I study of oral panobinostat alone and in combination with docetaxel in patients with castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 2010, 66, 181–189. [Google Scholar] [CrossRef]
- Wierstra, I.; Alves, J. FOXM1, a typical proliferation-associated transcription factor. Biol. Chem. 2007, 388, 1257–1274. [Google Scholar] [CrossRef]
- Briest, F.; Berg, E.; Grass, I.; Freitag, H.; Kaemmerer, D.; Lewens, F.; Christen, F.; Arsenic, R.; Altendorf-Hofmann, A.; Kunze, A. FOXM1: A novel drug target in gastroenteropancreatic neuroendocrine tumors. Oncotarget 2015, 6, 8185. [Google Scholar] [CrossRef]
- Laoukili, J.; Kooistra, M.R.; Brás, A.; Kauw, J.; Kerkhoven, R.M.; Morrison, A.; Clevers, H.; Medema, R.H. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat. Cell Biol. 2005, 7, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Laoukili, J.; Stahl, M.; Medema, R.H. FoxM1: At the crossroads of ageing and cancer. Biochim. Biophys. Acta (BBA) Rev. Cancer 2007, 1775, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Hamurcu, Z.; Ashour, A.; Kahraman, N.; Ozpolat, B. FOXM1 regulates expression of eukaryotic elongation factor 2 kinase and promotes proliferation, invasion and tumorgenesis of human triple negative breast cancer cells. Oncotarget 2016, 7, 16619. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Teng, M.; Liu, J.; Jin, D.; Wu, J.; Yan, D.; Fan, J.; Qin, X.; Tang, H.; Peng, Z. FOXM1 expression predicts the prognosis in hepatocellular carcinoma patients after orthotopic liver transplantation combined with the Milan criteria. Cancer Lett. 2011, 306, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.-Y.; Zhu, Z.-M.; Chen, L.-B.; Wang, J.-H.; Su, Q.-S.; Yang, J.-R.; Lin, Y.; Xue, L.-J.; Liu, X.-B.; Mo, X.-B. FOXM1 expression correlates with tumor invasion and a poor prognosis of colorectal cancer. Acta Histochem. 2012, 114, 755–762. [Google Scholar] [CrossRef]
- Zeng, J.; Wang, L.; Li, Q.; Li, W.; Björkholm, M.; Jia, J.; Xu, D. FoxM1 is up-regulated in gastric cancer and its inhibition leads to cellular senescence, partially dependent on p27kip1. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2009, 218, 419–427. [Google Scholar]
- de Moraes, G.N.; Delbue, D.; Silva, K.L.; Robaina, M.C.; Khongkow, P.; Gomes, A.R.; Zona, S.; Crocamo, S.; Mencalha, A.L.; Magalhães, L.M. FOXM1 targets XIAP and Survivin to modulate breast cancer survival and chemoresistance. Cell. Signal. 2015, 27, 2496–2505. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Fujiwara, Y.; Takahashi, T.; Nakamura, Y.; Takiguchi, S.; Nakajima, K.; Miyata, H.; Yamasaki, M.; Kurokawa, Y.; Mori, M. Overexpression of forkhead box M1 transcription factor (FOXM1) is a potential prognostic marker and enhances chemoresistance for docetaxel in gastric cancer. Ann. Surg. Oncol. 2013, 20, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Chiu, W.-T.; Huang, Y.-F.; Tsai, H.-Y.; Chen, C.-C.; Chang, C.-H.; Huang, S.-C.; Hsu, K.-F.; Chou, C.-Y. FOXM1 confers to epithelial-mesenchymal transition, stemness and chemoresistance in epithelial ovarian carcinoma cells. Oncotarget 2015, 6, 2349. [Google Scholar] [CrossRef] [Green Version]
- Gartel, A.L. FoxM1 inhibitors as potential anticancer drugs. Expert Opin. Ther. Targets 2008, 12, 663–665. [Google Scholar] [CrossRef] [PubMed]
- Adami, G.R.; Ye, H. Future roles for FoxM1 inhibitors in cancer treatments. Future Oncol. 2007, 3, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Park, M.H.; Kim, S.M. 3,3’-Diindolylmethane potentiates paclitaxel-induced antitumor effects on gastric cancer cells through the Akt/FOXM1 signaling cascade. Oncol Rep. 2015, 33, 2031–2036. [Google Scholar] [CrossRef] [Green Version]
- Yung, M.M.H.; Chan, D.W.; Liu, V.W.S.; Yao, K.-M.; Ngan, H.Y.-S. Activation of AMPK inhibits cervical cancer cell growth through AKT/FOXO3a/FOXM1 signaling cascade. BMC Cancer 2013, 13, 327. [Google Scholar] [CrossRef] [Green Version]
- Anne, M.; Sammartino, D.; Barginear, M.F.; Budman, D. Profile of panobinostat and its potential for treatment in solid tumors: An update. Onco Targets Ther. 2013, 6, 1613–1624. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.F.; Bendell, J.C.; Infante, J.R.; Spigel, D.R.; Thompson, D.S.; Yardley, D.A.; Greco, F.A.; Murphy, P.B.; Burris, H.A., III. A phase I study of panobinostat in combination with gemcitabine in the treatment of solid tumors. Clin. Adv. Hematol. Oncol. 2011, 9, 225–230. [Google Scholar]
- Bauer, S.; Hilger, R.A.; Grabellus, F.; Nagarajah, J.; Hoiczyk, M.; Reichardt, A.; Ahrens, M.; Pink, D.; Mühlenberg, T.; Scheulen, M.E. Phase I trial of panobinostat (P) and imatinib (IM) in patients with treatment-refractory gastrointestinal stromal tumors (GIST). J. Clin. Oncol. 2012, 30, 10032. [Google Scholar] [CrossRef]
- Gupta, M.; Han, J.; Stenson, M.; Wellik, L.; Witzig, T.E. Regulation of STAT3 by histone deacetylase-3 in diffuse large B-cell lymphoma: Implications for therapy. Leukemia 2012, 26, 1356–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Syn, N.L.-X.; Subhash, V.V.; Any, Y.; Thuya, W.L.; Cheow, E.S.H.; Kong, L.; Yu, F.; Peethala, P.C.; Wong, A.L.-A. Pan-HDAC inhibition by panobinostat mediates chemosensitization to carboplatin in non-small cell lung cancer via attenuation of EGFR signaling. Cancer Lett. 2018, 417, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, G. Effect of HDACi, Panobinostat, on Receptor Tyrosine Kinase-Induced Pathways in Glioblastoma. Master’s Thesis, Long Island University, The Brooklyn Center, MI, USA, 30 July 2014. [Google Scholar]
- Pietschmann, K.; Bolck, H.A.; Buchwald, M.; Spielberg, S.; Polzer, H.; Spiekermann, K.; Bug, G.; Heinzel, T.; Böhmer, F.-D.; Krämer, O.H. Breakdown of the FLT3-ITD/STAT5 axis and synergistic apoptosis induction by the histone deacetylase inhibitor panobinostat and FLT3-specific inhibitors. Mol. Cancer Ther. 2012, 11, 2373–2383. [Google Scholar] [CrossRef] [Green Version]
- Koo, C.-Y.; Muir, K.W.; Lam, E.W.-F. FOXM1: From cancer initiation to progression and treatment. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2012, 1819, 28–37. [Google Scholar] [CrossRef]
- Wang, Z.; Park, H.J.; Carr, J.R.; Chen, Y.-j.; Zheng, Y.; Tyner, A.L.; Costa, R.H.; Bagchi, S.; Raychaudhuri, P. FoxM1 in tumorigenicity of the neuroblastoma cells and renewal of the neural progenitors. Cancer Res. 2011, 71, 4292–4302. [Google Scholar] [CrossRef] [Green Version]
- Raychaudhuri, P.; Park, H.J. FoxM1: A master regulator of tumor metastasis. Cancer Res. 2011, 71, 4329–4333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Deshmukh, H.; Payton, J.E.; Dunham, C.; Scheithauer, B.W.; Tihan, T.; Prayson, R.A.; Guha, A.; Bridge, J.A.; Ferner, R.E. Array-based comparative genomic hybridization identifies CDK4 and FOXM1 alterations as independent predictors of survival in malignant peripheral nerve sheath tumor. Clin. Cancer Res. 2011, 17, 1924–1934. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.-T.; Wang, H.; Liang, L.-J.; Peng, B.-G.; Wu, Z.-F.; Chen, L.-Z.; Xue, L.; Li, Z.; Li, W. Overexpression of FOXM1 is associated with poor prognosis and clinicopathologic stage of pancreatic ductal adenocarcinoma. Pancreas 2012, 41, 629–635. [Google Scholar] [CrossRef]
- Xia, L.; Huang, W.; Tian, D.; Zhu, H.; Zhang, Y.; Hu, H.; Fan, D.; Nie, Y.; Wu, K. Upregulated FoxM1 expression induced by hepatitis B virus X protein promotes tumor metastasis and indicates poor prognosis in hepatitis B virus-related hepatocellular carcinoma. J. Hepatol. 2012, 57, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Wen, N.; Wang, Y.; Wen, L.; Zhao, S.-H.; Ai, Z.-H.; Wang, Y.; Wu, B.; Lu, H.-X.; Yang, H.; Liu, W.-C. Overexpression of FOXM1 predicts poor prognosis and promotes cancer cell proliferation, migration and invasion in epithelial ovarian cancer. J. Transl. Med. 2014, 12, 134. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qi, W.; Yao, R.; Tang, D.; Liang, J. Overexpressed transcription factor FOXM1 is a potential diagnostic and adverse prognostic factor in postoperational gastric cancer patients. Clin. Transl. Oncol. 2014, 16, 307–314. [Google Scholar] [CrossRef]
- Yao, S.; Fan, L.Y.-N.; Lam, E.W.-F. The FOXO3-FOXM1 axis: A key cancer drug target and a modulator of cancer drug resistance. Semin. Cancer Biol. 2018, 50, 77–89. [Google Scholar] [CrossRef]
- Costa, R.H. FoxM1 dances with mitosis. Nat. Cell Biol. 2005, 7, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, F.C.; O’sullivan, H. FOXM1 in sarcoma: Role in cell cycle, pluripotency genes and stem cell pathways. Oncotarget 2016, 7, 42792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, N.; Chakraborty, S.; Nag, A. Biology of FOXM1 and its emerging role in cancer therapy. J. Proteins Proteom. 2014, 5, 1–24. [Google Scholar]
- Jia, L.; Jin, H.; Zhou, J.; Chen, L.; Lu, Y.; Ming, Y.; Yu, Y. A potential anti-tumor herbal medicine, Corilagin, inhibits ovarian cancer cell growth through blocking the TGF-β signaling pathways. BMC Complement. Altern. Med. 2013, 13, 33. [Google Scholar] [CrossRef] [Green Version]
- Harkness, T.A.A. Mechanistic insights into aging, cell-cycle progression, and stress response. Front. Physiol. 2012, 3, 183. [Google Scholar]
- Kato, M.; Yuan, H.; Xu, Z.-G.; Lanting, L.; Li, S.-L.; Wang, M.; Hu, M.C.-T.; Reddy, M.A.; Natarajan, R. Role of the Akt/FoxO3a pathway in TGF-β1–mediated mesangial cell dysfunction: A novel mechanism related to diabetic kidney disease. J. Am. Soc. Nephrol. 2006, 17, 3325–3335. [Google Scholar] [CrossRef] [Green Version]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, N.-R.; Kim, D.-Y.; Jin, H.; Meng, R.; Chai, O.H.; Kim, S.-H.; Park, B.-H.; Kim, S.M. Inactivation of the Akt/FOXM1 Signaling Pathway by Panobinostat Suppresses the Proliferation and Metastasis of Gastric Cancer Cells. Int. J. Mol. Sci. 2021, 22, 5955. https://doi.org/10.3390/ijms22115955
Lee N-R, Kim D-Y, Jin H, Meng R, Chai OH, Kim S-H, Park B-H, Kim SM. Inactivation of the Akt/FOXM1 Signaling Pathway by Panobinostat Suppresses the Proliferation and Metastasis of Gastric Cancer Cells. International Journal of Molecular Sciences. 2021; 22(11):5955. https://doi.org/10.3390/ijms22115955
Chicago/Turabian StyleLee, Na-Ri, Da-Yeah Kim, Hua Jin, Ruoyu Meng, Ok Hee Chai, Seong-Hun Kim, Byung-Hyun Park, and Soo Mi Kim. 2021. "Inactivation of the Akt/FOXM1 Signaling Pathway by Panobinostat Suppresses the Proliferation and Metastasis of Gastric Cancer Cells" International Journal of Molecular Sciences 22, no. 11: 5955. https://doi.org/10.3390/ijms22115955